Projekt A02: Mechanismen des Zystenwachstums bei ADPKD
Im Teilprojekt A02 beschäftigen wir uns mit der Pathophysiologie der autosomaldominanten polyzystischen Nierenerkrankung (ADPKD). Mit einer Häufigkeit von etwa 1:1000 ist ADPKD die häufigste monogenetisch verursachte Nierenerkrankung und zählt zu den wichtigsten Gründen für die Notwendigkeit einer Nierenersatztherapie.
Pathophysiologie der autosomaldominanten polyzystischen Nierenerkrankung (ADPKD)
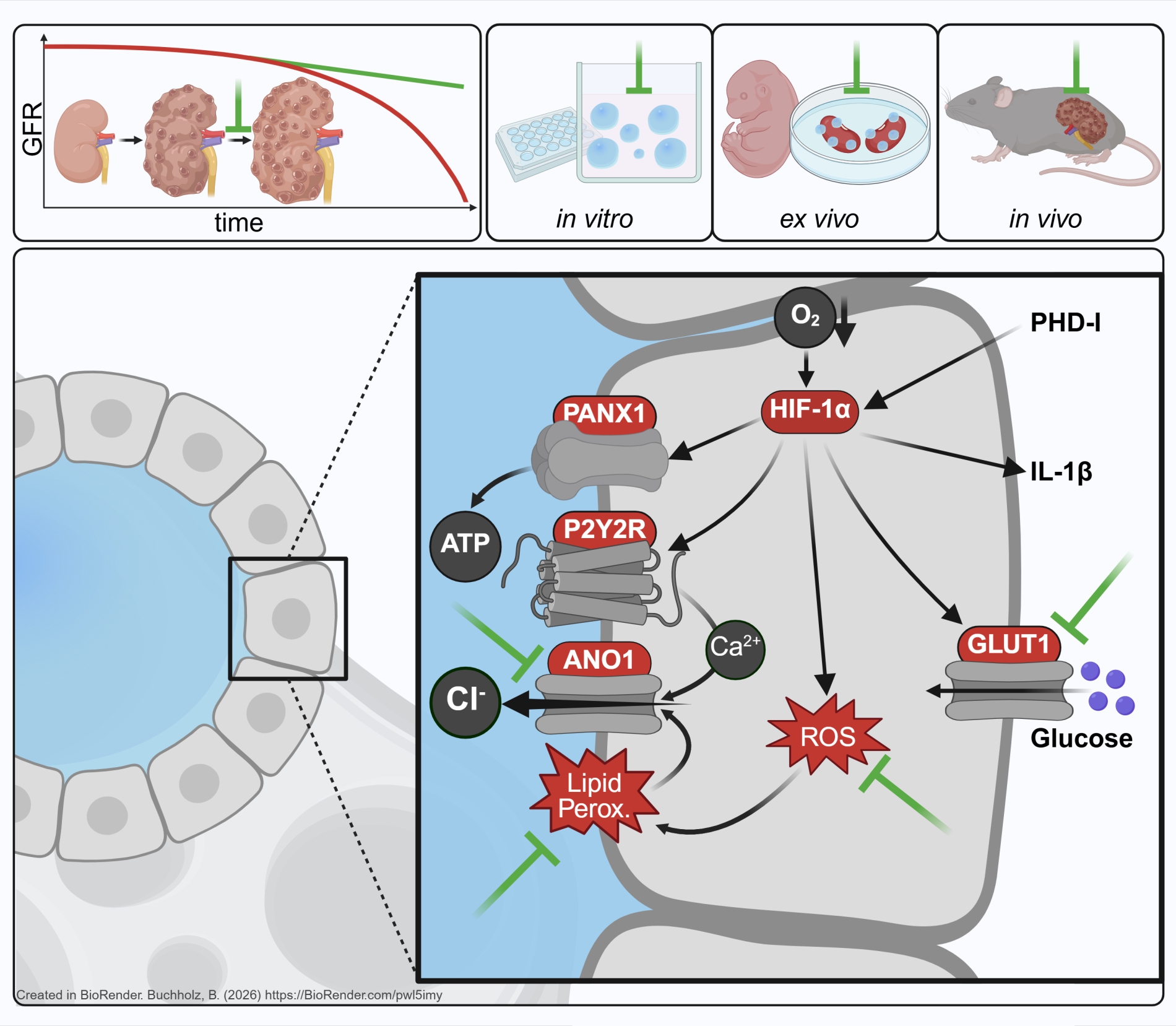
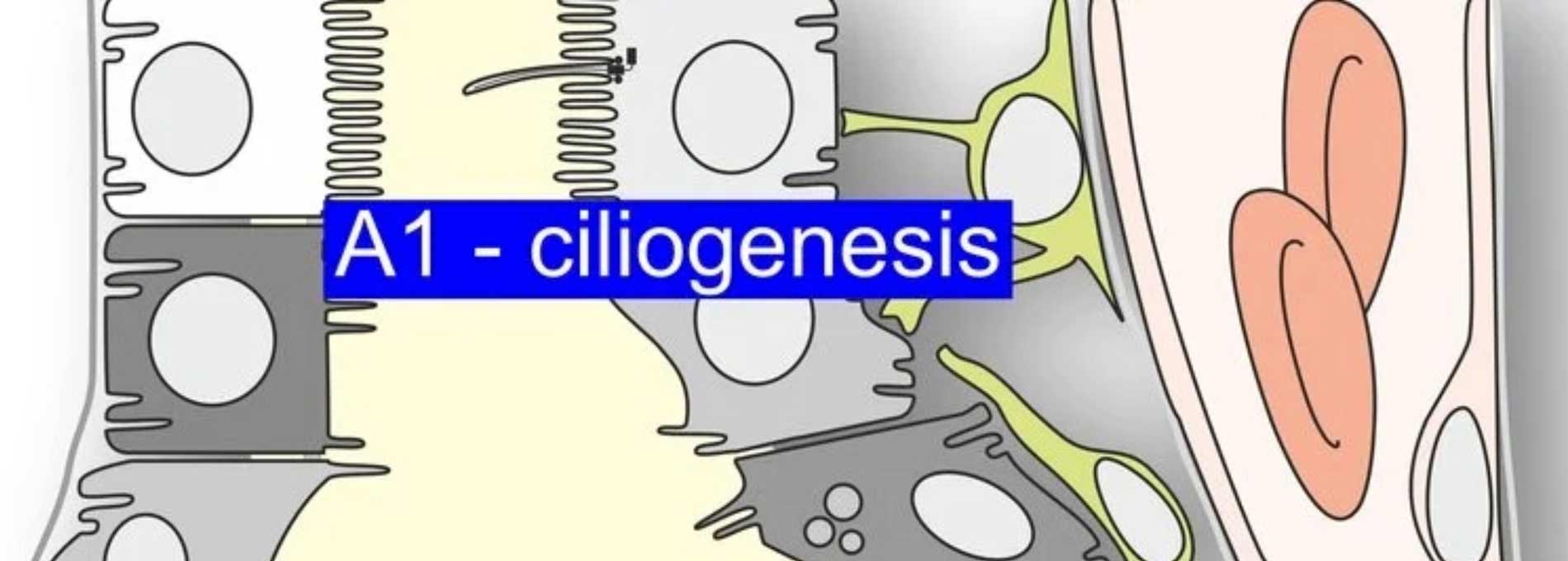
Ursächlich ist meist eine Mutation im PKD1Gen, das für Polycystin1 kodiert, gefolgt von Mutationen im PKD2Gen, das für Polycystin2 (TRPP2) kodiert. Klinisch ist ADPKD durch die Bildung zahlreicher Zysten (flüssigkeitsgefüllter Hohlräume) in beiden Nieren gekennzeichnet. Die Zysten entstehen aus einzelnen Nephronen und Sammelrohren und wachsen über Jahrzehnte kontinuierlich weiter. Dies führt zu einer zunehmenden Kompression gesunder Nierenanteile und damit zu einem progredienten Verlust der Nierenfunktion. Die Verbindung zwischen den Genmutationen und der Entstehung der Zysten ist bislang nur unzureichend verstanden. Wir fokussieren uns daher darauf, die Mechanismen des Zystenwachstums zu entschlüsseln, um pharmakologisch adressierbare Ziele zu identifizieren, die das Zystenwachstum stoppen und so die Nierenfunktion erhalten könnten.
Hemmung des Zystenwachstums über Ionenkanäle
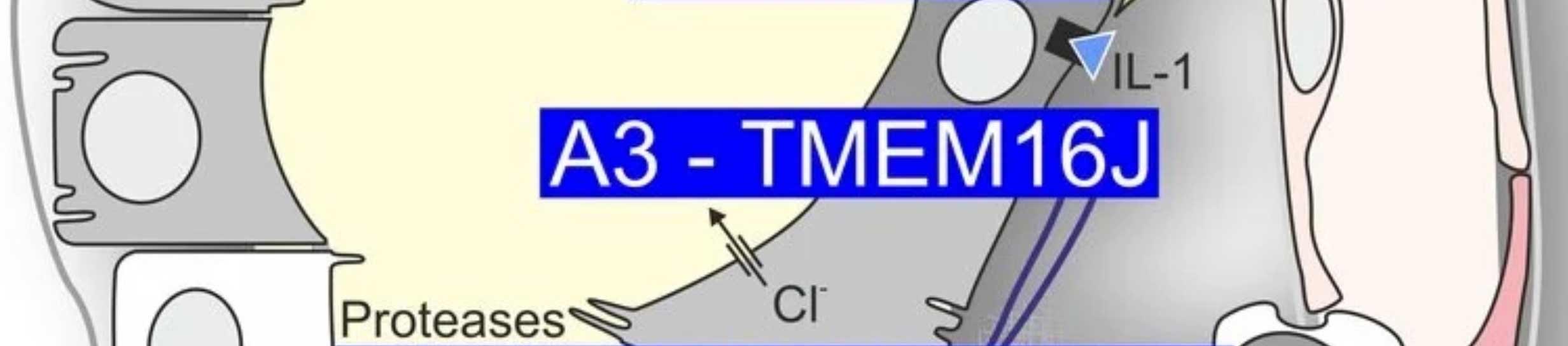
Im Rahmen der bisherigen Förderung im SFB1350 bzw. TRR374 konnten wir zeigen, dass das progrediente Zystenwachstum zu lokaler Hypoxie und konsekutiv zur Expression des hypoxieinduzierten Transkriptionsfaktors HIF1α im Zystenepithel führt. HIF1α verstärkt das Zystenwachstum, indem es die Chloridsekretion, und damit auch den Wassertransport, in das Zystenlumen signifikant erhöht. Entgegen früheren Annahmen konnten wir in unseren Modellen zeigen, dass der Chloridkanal CFTR hierbei eine eher untergeordnete Rolle spielt, während der kalziumaktivierte Chloridkanal TMEM16A (Anoctamin1; ANO1) maßgeblich zur Sekretion beiträgt. Spezifische Inhibitoren von TMEM16A sowie bereits für andere Indikationen zugelassene Medikamente wie Benzbromaron und Niclosamid, die ebenfalls TMEM16A hemmen, reduzierten das Zystenwachstum in vitro, ex vivo und in vivo signifikant.
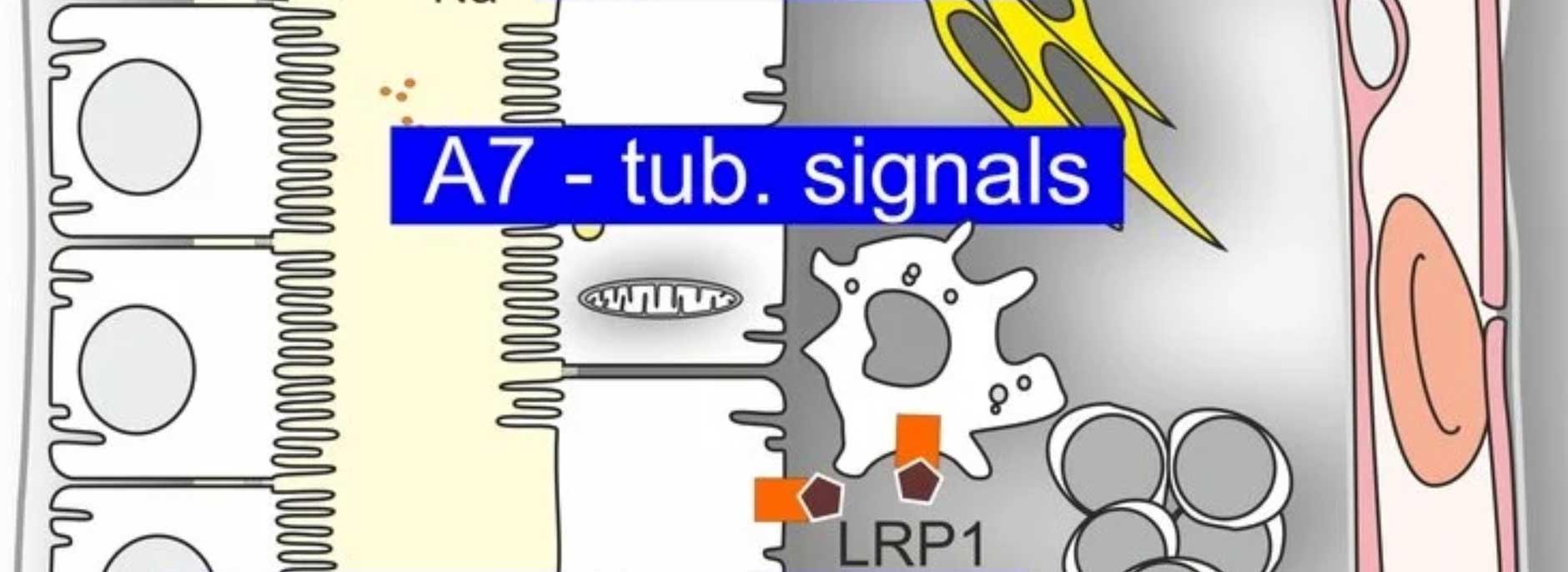
Ergänzend fanden wir heraus, dass die Aktivierung von TMEM16A durch ATP, das im Zystenlumen akkumuliert, über den purinergen Rezeptor P2Y2R vermittelt wird, der somit ebenfalls ein potenzielles pharmakologisches Target darstellt. Passend zur Rolle von HIF1α konnten wir zeigen, dass P2Y2R ein Zielgen von HIF1α ist und dass der Transport von ATP in das Zystenlumen ebenfalls durch HIF1α gesteigert wird. Neben ATP führt auch eine für das Zystenepithel charakteristische verstärkte Lipidperoxidation zu einer erhöhten Offenwahrscheinlichkeit von TMEM16A. Pharmakologische Interventionen, die oxidativen Stress und damit Lipidperoxidation reduzieren, führten ebenfalls zu vermindertem Zystenwachstum.
Kombination von therapeutischen Strategien für besseres Therapieansprechen
Da HIF1α ein zentraler und essenzieller Regulator zahlreicher zellulärer Prozesse ist, eignet es sich selbst nicht als therapeutisches Ziel. Dennoch ist es klinisch relevant, da wir zeigen konnten, dass der Einsatz von ProlylhydroxylaseInhibitoren, die zur Behandlung der renalen Anämie zugelassen sind, das Zystenwachstum verstärken kann. Ausgehend von der vielseitigen Rolle von HIF1α untersuchten wir neben Effekten auf die Chloridsekretion auch mögliche Einflüsse auf Inflammation und Metabolismus und konnten dabei weitere potenzielle, bislang noch unpublizierte, Targets identifizieren.
Unser nächstes Ziel ist es nun, pharmakologische Strategien gezielt zu kombinieren, um parallel in unterschiedlichen Signalwegen zu intervenieren und so den größtmöglichen Effekt auf das Zystenwachstum zu erzielen. Gleichzeitig soll die Konzentration der einzelnen Substanzen möglichst niedrig gehalten werden, um potenzielle Nebenwirkungen, insbesondere im Rahmen einer langfristigen Therapie, zu minimieren.

Publikationen (Auswahl)
- Kraus, A., Skoczynski, K., Brötsch, M., Burzlaff, N., Leipziger, J., Schiffer, M., Büttner‑Herold, M., & Buchholz, B. (2024). P2Y2R and Cyst Growth in Polycystic Kidney Disease. J. Am. Soc. Nephrol.
https://doi.org/10.1681/ASN.0000000000000416 - Grampp, S., Kraus, A., Skoczynski, K., Schiffer, M., Krüger, R., Naas, S., Schödel, J., & Buchholz, B. (2023). Hypoxia induces polycystin‑1 expression in the renal epithelium. R. Soc. Open Sci. 10(5), 220992.
https://doi.org/10.1098/rsos.220992 - Scholz, J. K., Kraus, A., Lüder, D., Skoczynski, K., Schiffer, M., Grampp, S., Schödel, J., & Buchholz, B. (2022). Loss of Polycystin‑1 causes cAMP‑dependent switch from tubule to cyst formation. iScience 25(6), 104359.
https://doi.org/10.1016/j.isci.2022.104359 - Talbi, K., Cabrita, I., Kraus, A., Hofmann, S., Skoczynski, K., Kunzelmann, K., Buchholz, B., & Schreiber, R. (2021). The chloride channel CFTR is not required for cyst growth in an ADPKD mouse model. FASEB J. 35(10), e21897.
https://doi.org/10.1096/fj.202100843R - Buchholz, B., & Eckardt, K.-U. (2020). Role of oxygen and the HIF‑pathway. Cell. Signal. 109524.
https://doi.org/10.1016/j.cellsig.2020.109524 - Cabrita, I., Buchholz, B., Schreiber, R., & Kunzelmann, K. (2020). TMEM16A drives renal cyst growth by augmenting Ca²⁺ signaling in M1 cells. J. Mol. Med. 98(5), 659–671.
https://doi.org/10.1007/s00109-020-01894-y - Cabrita, I., Kraus, A., Scholz, J. K., Skoczynski, K., Schreiber, R., Kunzelmann, K., & Buchholz, B. (2020). Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat. Commun. 11, 4320.
https://doi.org/10.1038/s41467-020-18104-5 - Schreiber, R., Buchholz, B., Kraus, A., Schley, G., Scholz, J., Ousingsawat, J., & Kunzelmann, K. (2019). Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 30(2), 228–242.
https://doi.org/10.1681/ASN.2018010039 - Kraus, A., Peters, D. J. M., Klanke, B., Weidemann, A., Willam, C., Schley, G., Kunzelmann, K., Eckardt, K.-U., & Buchholz, B. (2018). HIF‑1α promotes cyst progression in a mouse model of autosomal dominant polycystic kidney disease. Kidney Int. 94(5), 887–899.
https://doi.org/10.1016/j.kint.2018.06.008